What is band theory? In the 1970s, the general problem of the electronic structure of crystalline solids was undergoing a period of rapid theoretical and methodological development. The crystal structures possible in the solid state were fully understood, and very many were known experimentally from x-ray or neutron diffraction. The corresponding electronic structure problem was framed in terms of a 1-electron Schrödinger equation containing a potential with periodic, translational, symmetry. Even though it is a gross simplification to treat the electrons as independent particles when they are clearly strongly interacting, it presented a serious enough computational challenge at that time. Many methods were developed for solving this “band theory” problem for realistic potentials: plane-wave-based methods (orthogonalised – OPW, augmented – APW); pseudopotential methods; multiple scattering methods (KKR); and so on [1]. Each method tended to be used for different classes of materials: simple metals, transition metals, semiconductors etc. The eigenvalue spectrum of the periodic Hamiltonian is the energy band structure, ie the set of energy levels En (k) as a function of the cryst
What is band theory? In the 1970s, the general problem of the electronic structure of crystalline solids was undergoing a period of rapid theoretical and methodological development. The crystal structures possible in the solid state were fully understood, and very many were known experimentally from x-ray or neutron diffraction. The corresponding electronic structure problem was framed in terms of a 1-electron Schrödinger equation containing a potential with periodic, translational, symmetry. Even though it is a gross simplification to treat the electrons as independent particles when they are clearly strongly interacting, it presented a serious enough computational challenge at that time. Many methods were developed for solving this “band theory” problem for realistic potentials: plane-wave-based methods (orthogonalised – OPW, augmented – APW); pseudopotential methods; multiple scattering methods (KKR); and so on [1]. Each method tended to be used for different classes of materials: simple metals, transition metals, semiconductors etc. The eigenvalue spectrum of the periodic Hamiltonian is the energy band structure, ie the set of energy levels En (k) as a function of the cryst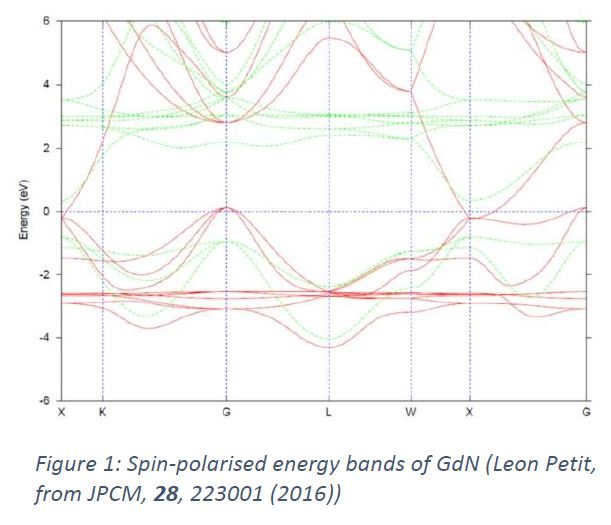 al momentum k. This is frequently pretty complicated (see Figure 1); hence its nickname “spaghetti” among band theorists.
al momentum k. This is frequently pretty complicated (see Figure 1); hence its nickname “spaghetti” among band theorists.
But perhaps the main point to note about early band structure calculations is that they tended to agree surprisingly well with experimental data, for example from Fermi surface and angleresolved photoemission measurements. It seemed that the amount of physics one could get out of these calculations was quite impressive, especially given the potentials used in these early days (mostly derived from atomic calculations by physically motivated schemes – ie cookery). Here was evidence that solving the 1-electron Schrödinger equation was going to be an essential foundation for more accurate methods based on more rigorous theory. And so it has proved. Nowadays (nearly) every band structure calculation is derived from density functional theory (DFT), a self-consistent field procedure whose theoretical foundations and limits are well understood. In particular, DFT gives the ground state energetics of a system rigorously and now reached levels of accuracy that underpin the emerging field of computational materials science. Such calculations are also the starting point for modern discussions of more spectacular many-electron effects in solids – magnetism, superconductivity, most recently “topological states” of matter [2].
 Now rewind to the UK in the 1970s. Several solid state theory groups were interested in various aspects of electronic structure – Cambridge, Bristol, Imperial College, Leeds, etc. The usefulness of numerical calculations was becoming apparent, in the USA and some European countries, but the capability to do serious band structure calculations, especially self-consistent ones, was only embryonic in the UK. Balazs Gyorffy and Malcolm Stocks in the Bristol physics department had the idea of making a collaborative attack on this issue, aiming to develop and import leading band theory methods and codes and make them available to the UK research community. They mounted Figure 1: Spin-polarised energy bands of GdN (Leon Petit, from JPCM, 28, 223001 (2016)) a campaign to establish a “Band Theory Project” of which this letter to the community was a part. When I recently came across the letter buried in some old files, it brought back to me the passion with which Balazs and Malcolm pursued this initiative and the conversations they stimulated in the community. At the time, Balazs was a young lecturer in John Ziman’s theory group in Bristol, and Malcolm was, I think, a postdoc. Balazs was a proper theoretician, full of ideas and energy, and the most stimulating research collaborator one could wish for. Malcolm’s great strength was the ability, pretty rare at that time, to put theories into numerical practice. Together, Balazs and Malcolm were a formidable team. Walter Temmerman and I were PhD students in Bristol before coming to Daresbury, and we benefitted from being around Balazs and Malcolm in so many ways throughout our careers. Balazs passed away in 2012 after a notable scientific and intellectual life in Bristol [3], and is sorely missed. Malcolm went on to a very successful career in Oak Ridge National Laboratory, becoming one of the leading band theorists in the USA, and is still going strong.
Now rewind to the UK in the 1970s. Several solid state theory groups were interested in various aspects of electronic structure – Cambridge, Bristol, Imperial College, Leeds, etc. The usefulness of numerical calculations was becoming apparent, in the USA and some European countries, but the capability to do serious band structure calculations, especially self-consistent ones, was only embryonic in the UK. Balazs Gyorffy and Malcolm Stocks in the Bristol physics department had the idea of making a collaborative attack on this issue, aiming to develop and import leading band theory methods and codes and make them available to the UK research community. They mounted Figure 1: Spin-polarised energy bands of GdN (Leon Petit, from JPCM, 28, 223001 (2016)) a campaign to establish a “Band Theory Project” of which this letter to the community was a part. When I recently came across the letter buried in some old files, it brought back to me the passion with which Balazs and Malcolm pursued this initiative and the conversations they stimulated in the community. At the time, Balazs was a young lecturer in John Ziman’s theory group in Bristol, and Malcolm was, I think, a postdoc. Balazs was a proper theoretician, full of ideas and energy, and the most stimulating research collaborator one could wish for. Malcolm’s great strength was the ability, pretty rare at that time, to put theories into numerical practice. Together, Balazs and Malcolm were a formidable team. Walter Temmerman and I were PhD students in Bristol before coming to Daresbury, and we benefitted from being around Balazs and Malcolm in so many ways throughout our careers. Balazs passed away in 2012 after a notable scientific and intellectual life in Bristol [3], and is sorely missed. Malcolm went on to a very successful career in Oak Ridge National Laboratory, becoming one of the leading band theorists in the USA, and is still going strong.
 One final point. The Band Theory Project as such never got off the ground. But the proposal coincided with the establishment of the Theory and Computational Science Division and of the Collaborative Computational Projects (CCPs) at Daresbury, under the leadership of Phil Burke and John Pendry. In 1980, CCP9, the CCP on “The Electronic Structure of Solids”, was funded, with Balazs as its first Chair and Walter as its first secretary. The rest is history. CCP9 is still in full operation today, naturally with different theories, codes and scientific interests, but with the same philosophy and ethos expressed so eloquently in the Band Theory Project letter of Balazs and Malcolm all those years ago. Smart guys!
One final point. The Band Theory Project as such never got off the ground. But the proposal coincided with the establishment of the Theory and Computational Science Division and of the Collaborative Computational Projects (CCPs) at Daresbury, under the leadership of Phil Burke and John Pendry. In 1980, CCP9, the CCP on “The Electronic Structure of Solids”, was funded, with Balazs as its first Chair and Walter as its first secretary. The rest is history. CCP9 is still in full operation today, naturally with different theories, codes and scientific interests, but with the same philosophy and ethos expressed so eloquently in the Band Theory Project letter of Balazs and Malcolm all those years ago. Smart guys!

[1] “Electronic Structure”, Richard M Martin (Cambridge University Press, 2012)
[2] “Topological Insulators and Topological Superconductors”, B A Bernevig with T L Hughes (Princeton University Press, 2013)
[3] “Correlation and disorder, in honour of Balazs Gyorffy”, special issue of J Phys Condensed Matter vol 26 (2014)
 The Gyorffy-Stocks letter.pdf
The Gyorffy-Stocks letter.pdf