Jakub Adamski
University of Edinburgh
| Energy Efficient Quantum Computing Simulations
As we are entering the era when quantum advantage becomes viable, it is especially important to push the boundaries of classical simulations of quantum computing. It involves running exponentially complex algorithms, so the use of high-performance computing is essential and entails huge energy consumption. The simulation can be performed via state vector evolution or by contracting a tensor network of matrix product states and operators. Each method offers different advantages, and allows potential optimisations to save energy. Various benchmarks have been set up and run on ARCHER2 to determine the most economical approach. It was found that by downclocking the CPU, a state vector simulation can consume up to 30% less energy. On the other hand, tensor networks proved exponentially more efficient when the entanglement was limited. The goal of this poster is to present and explain the benchmarking results, and encourage greener HPC use when simulating quantum computing.
|
| Bruno Camino University College London
| Applications of quantum computing for quantum chemistry Quantum chemistry has been predicted to be one of the first fields to benefit from the development of quantum computing. In this work we explore applications of quantum annealing for the study of solid solutions. These materials are of great interest for energy store applications and simulating their properties with classical computers is particularly challenging because of the large configuration space to explore. Using vacancies of graphene as a model system, we show how quantum annealers can be used to tackle these type of problems.
|
Shayantan Chaudhuri
University of Warwick
| Long-range dispersion-inclusive machine learning potentials for hybrid organic-inorganic interfaces The computational prediction of the structure and stability of hybrid organic–inorganic interfaces provides important insights into the measurable properties of electronic thin film devices and catalyst surfaces, and plays an important role in their rational design. However, the rich diversity of molecular configurations and the important role of long-range interactions in such systems make it difficult to use machine learning potentials (MLPs) to facilitate structure exploration that would otherwise require computationally expensive electronic structure calculations. We present an ML approach that enables fast, yet accurate, structure optimisations by combining two different types of deep neural networks trained on high-level electronic structure data for gold nanoclusters on diamond (110) surfaces.
|
Kenneth Chinembiri
University of Sheffield
| An Immersed Boundary Method for the DNS Solver CHAPSIM CHAPSim 2.0 is a Direct Numerical Simulation code developed by the Collaborative Computational Project – Nuclear Thermal Hydraulics (CCP-NTH) as an open-source UK nuclear community code. The solver is fast, efficient, and capable of simulating turbulent thermal flows with strong physical property variation. This paper discusses the methodology and validation of an Immersed Boundary Method (IBM) for complex geometries in the solver CHAPSim 2.0. When adopting this method, the effect of the solid body to flow field is mimicked by introducing a forcing term to the governing momentum equations of the CFD solver. The forcing term allows the user to impose a desired target velocity at the grid nodes of the complex solid boundary and is computed courtesy of the direct forcing approach. This function enables CHAPSim2 to simulate flow over arbitrary geometry without complicated grid generation process.
|
Asa Hopkins
University of Strathclyde
| Introducing Incoherence to Artificial Neural Networks Artificial neural networks (NNs) at their core are an attempt to emulate the biological NNs found in the brains of animals, and can accomplish tasks with lower energy consumption than more traditional computing methods.However, there are still ways that artifical NNs fall short of their biological counterparts. Most artificial NNs are made up of layers of nodes, with edges only being formed between adjacent layers. This kind of strict ordering is not seen in nature and the existence of edges connecting non-adjacent layers is important to the stability of larger natural systems, such as food chains and metabolic pathways. The extent to which this strict layering is broken is known as the trophic incoherence.This work investigates methods of adding trophic incoherence to artificial NNs, and the effectsdoing so has on the convergence speed during training (fast convergenceis more energy efficient) and the accuracy after training is completed.
|
Lara Janiurek
University of Strathclyde
| Using Machine Learning Techniques to Determine Photometric Redshifts for Gravitational wave Cosmology The inference of the Hubble constant using gravitational waves has allowed for a new way for the expansion of the universe to be probed, which may shed light on the current Hubble tension. Galaxy redshift surveys are a required for the application of these dark sirens. Photometric redshift surveys contain significant errors and spectroscopic redshifts are much more energy intensive than simply using an algorithm to estimate these values. Here, the random forest (RF) algorithm GALPRO is implemented to generate photometric redshift posteriors. GALPRO is calibrated using a truth dataset, which is successful, meaning it is useful when presented with an incomplete survey with missing redshift values. Analysis suggests that the redshift posterior distributions are non-Gaussian. Tests were run which determined that training and testing datasets must overlap by least 90% in range to give accurate results. However, the algorithm failed when the training and testing datasets came from different surveys meaning there is some underlying fundamental difference in galaxy surveys that must be recognised when using RFs.
|
Harriet Jones
STFC / University of Chester
| Validation and Application of Lagrangian Stochastic Methods for Indoor Air Quality This STFC Air Quality Network (SAQN) project uses EDF’s computational fluid dynamics software Code_Saturne to model the dispersion of a key hazardous aerial pollutant, particulate matter (PM), during cooking experiments within a test house. This is done via the implementation of Code_Saturne’s Lagrangian Particle Tracking module. The basis for the model is the EPSRC funded DOMestic Systems Technology InCubator (DOMESTIC) test house, a controlled environment designed to simulate a full-scale kitchen/diner and bathroom. Early results indicate that the model appears to effectively replicate the evolution of PM2.5 (particulate matter with a diameter of 2.5 microns or less) during cooking episodes, and further experimental validation results are pending.
|
Emanuele Marsili
University of Bristol
| A Theoretical Perspective on the Actinic Photochemistry of 2-hydroperoxypropanal Determining the chemical composition of the Earth's troposphere and its evolution over time is crucial for shaping the political and societal decisions regarding global warming. Presently used chemical mechanism models - encompassing experimental and theoretical data for many ground-state reactions of volatile organic compounds (VOCs) - allow estimating the outcomes of VOCs reactions. Interestingly though, the role of light-induced, excited-state processes is still largely unexplored and photochemical reactions of transient VOCs are mostly neglected in predictive atmospheric models. 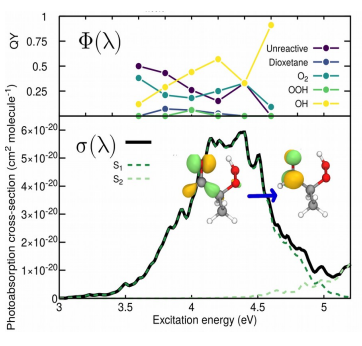 One important family of VOCs is the α-hydroperoxycarbonyls. Since experimental studies on these transient molecules are hardly feasible, we have employed high-level quantum chemical methods to fully characterize the photochemistry of the 2-hydroperoxypropanal (2-HPP) [1]. Using the nuclear ensemble approach we calculated the photo-absorption cross-section (σ(λ)) [2] while we resorted to nonadiabatic molecular dynamics to determine the wavelength-dependent photolysis quantum yield (Φ(λ)). These two ingredients, together with the solar actinic flux (F(λ)), allow us to predict the photolysis rate constant J, a crucial piece of information required by predictive chemical mechanism models. [1] Marsili E., et al. The Journal of Physical Chemistry A 2022, 126, 5420–5433 [2] Prlj A., Marsili E., et al., ACS Earth and Space Chemistry 2022, 6, 207-217 |