Shapespyer is a Python
toolchain and associated workflows, being developed by
Dr Andrey Brukhno (SCD), for carrying out computational
simulations of molecular nanostructures found in soft matter and biomolecular systems. Taking any number of sample (template) molecules as inputs, it can automatically generate larger multi-component structures that can be used as inputs for molecular dynamics simulations carried out on any computing platform. The results from these calculations include equilibrated structures and trajectories that can be analysed and compared with results from small angle scattering (e.g. SAS, SANS, SAXS) experiments.
For user's convinience, template molecules' 3D structure, i.e. coordinates, can be either (i) taken from an existing coordinate file, e.g. .gro or .xyz, or (ii) generated based on popular SMILES-string notation encoding the chemical structure of a molecule. The molecular aggregates that can be created, simulated and analysed include:
- flat disks (rings)
- balls (micelles and vesicles)
- stacks (cylinders, rods, worm-like structures)
- aggregates arranged on 3D lattice(s)
- bicontinuous 3D structures based on Schwarz minimal surfaces
The diagram to the right illustrates the stages in a general simulation workflow. Below exemplar snapshots are also shown for initial, intermediate and final structures obtained from an automated initialisation workflow in the case of SDS and CTAB surfactant micellar solution. The template molecule coordinates are taken from .pdb, .xyz or .gro (Gromacs) files.
Shapespyer automates the setup and batched runs of molecular dynamics (MD) simulations based on the force fields available in Gromacs package, e.g. CHARMM, OPLS, GROMOS, with an optional initialisation step of adding ionic species and solvent.
Specialised batch shell scripts (in Bash environment) are provided to
perform equilibration and production simulations, with optional post-simulation
analyses, on virtually any high performance parallel execution platform (HPC),
including standard CPU systems (e.g. ARCHER2)
and hybrid accelerated CPU/GPU systems (e.g. Baskerville). The
supported analyses on the simulation results include: (i) cluster classification
and size distribution analyses, (ii) radii of gyration and moments of inertia, (iii)
hydration layer and cavity occupation analyses, the results of which can be
compared with experimental data (where those are available). Background
In the domain of soft matter and biomolecular simulation, the task of preparing complex molecular structures of interest is both challenging and tedious. The naive approach using an initial configuration with randomised positions of solute molecules is most often fruitless due to the complexity of the (unknown) pathways that lead to self-assembly of any particular shape of a molecular aggregate. Unfortunately, pre-equilibration into and further relaxation of a specific molecular complex can easily require simulation times way beyond what is reachable in a practical simulation scenario. Therefore, it is vital to have efficient tools facilitating the generation and initial equilibration of pre-assembled molecular complexes with predetermined composition and structure. This is exactly the purpose of the Shapespyer package.
Shapespyer package has been created by
Dr Andrey Brukhno for the Ada Lovelace Centre (ALC) project '
Initial Trajectory Generation for Soft Matter Molecular Dynamics Simulations` between SCD (Computational Chemistry Group), ISIS and Diamond.
It is available under an open-source BSD-3 licence.
The Python tools, runscripts and documentation can be downloaded from Shakespyer's
repository on GitLab.
Project partners
- Michael Seaton, John Purton (Computational Chemistry, SCD)
- James Doutch (ISIS Neutron and Muon Source)
- Tim Snow (Diamond)
- Hanna Barriga (Karolinska Institutet, Sweden)
- Margaret Holme (Chalmers, Sweden)
- Karen Edler (Lund University)
- Tom Headen, H. Bindu Kolli (Disordered Materials Group, ISIS Neutron and Muon Source)
Workflow examples
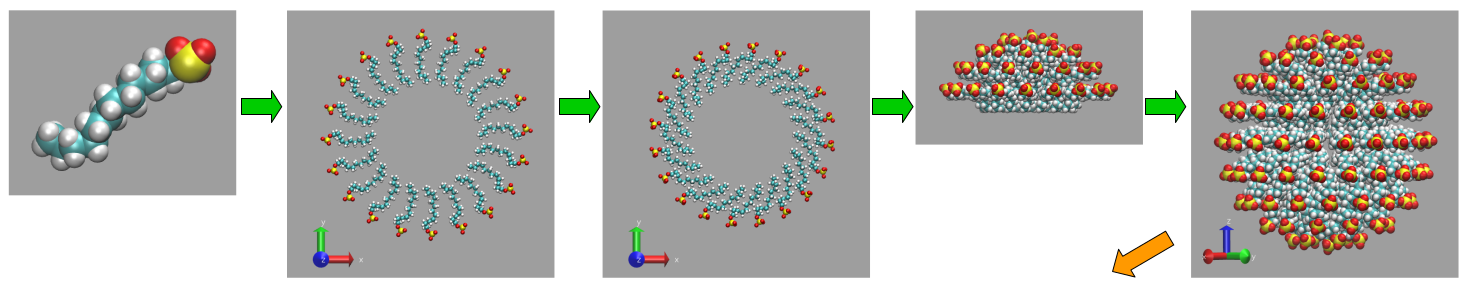


Example structures generated by Shapespyer from a single molecule of SDS (sodium dodecyl sulphate).
Bottom-right panel: Bicontinuous cubic phase with 50/50 mix of SDS and CTAB surfactants.
Largest cluster of CTAB (cetyl-trimethyl ammonium bromide) obtained from an MD simulation orchestrated by Shapespyer
and the results of post-simulation analysis for the radius of gyration and its primary components as a function of time.