Drs Ivan Scivetti and Gilberto Teobaldi (of the Scientific Computing Department -SCD) working with fellow scientists at the University of Liverpool and the National Tsing Hua University of Taiwan, developed a new research methodology combining high-resolution electrochemical measurements with atomistic simulations to derive unprecedented insights of the insertion of ions within battery (i.e. electro-active) materials.
Drs Ivan Scivetti and Gilberto Teobaldi (of the Scientific Computing Department -SCD) working with fellow scientists at the University of Liverpool and the National Tsing Hua University of Taiwan, developed a new research methodology combining high-resolution electrochemical measurements with atomistic simulations to derive unprecedented insights of the insertion of ions within battery (i.e. electro-active) materials.
Problem
Reversible ion-intercalation is one of the fundamental processes for electrochemical energy storage and conversion. In electrolyte ion batteries, this process involves the exchange of ions between the electrolyte and the host structure induced by charge/discharge voltage cycles. Repeated cycling eventually leads to battery degradation and loss of reversibility. This degradation occurs at different rates depending on the electrolytic ion species and the material. Unravelling the reasons behind degradation is key to developing high performing, safer and sustainable electro-active materials. This requires an in-depth understanding of the materials used, starting from the changes induced in their composition and structure following ion-intercalation, as well as any possible phase transformation.
Background
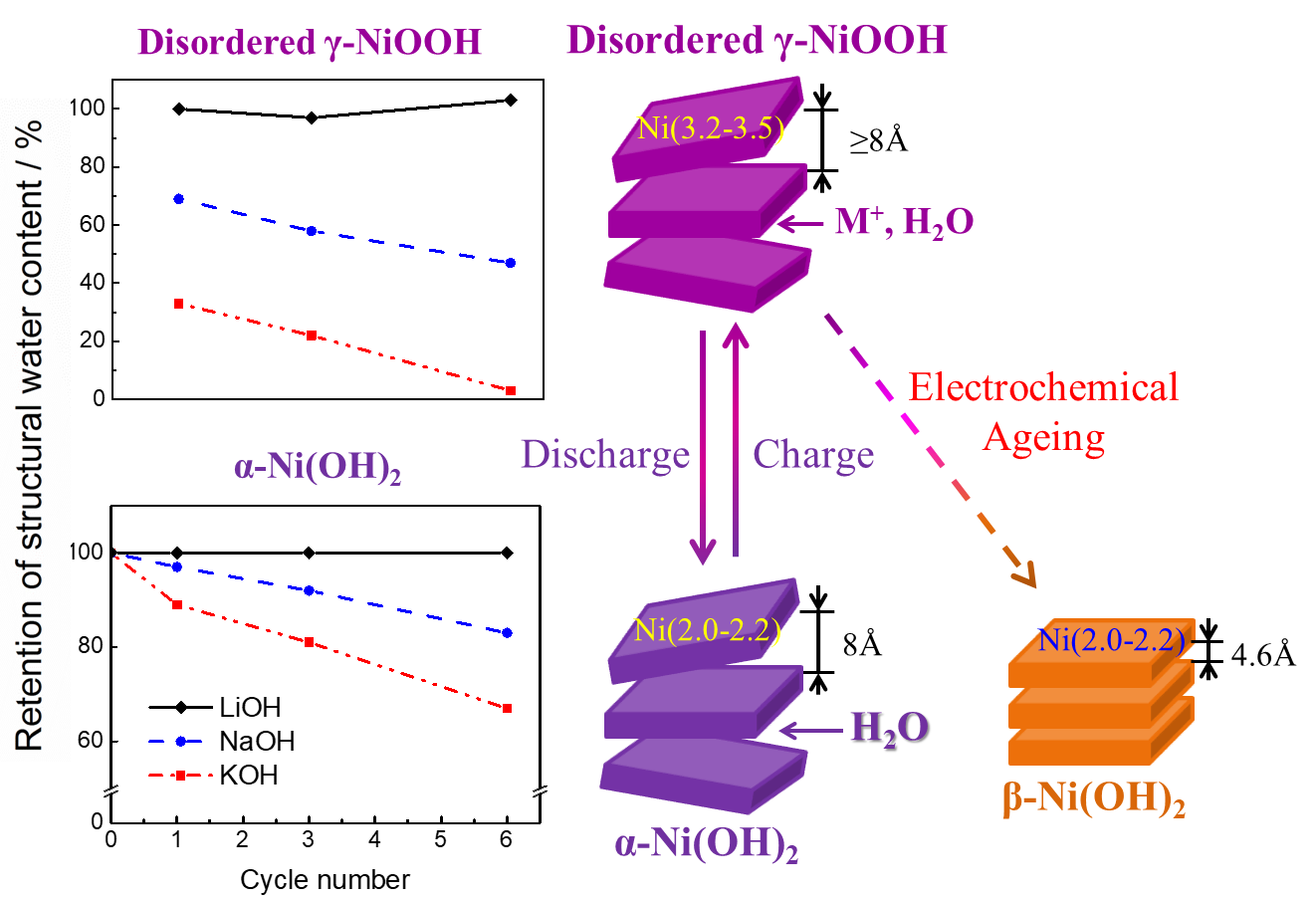 A typical electro-active material is nickel hydroxide –Ni(OH)2 comprising a layered structure with intercalated water, forming gaps or cavities between the layers (see page image). When in contact with an electrolyte containing ions such as K+, Na+ or Li+ the application of a voltage charge causes the ions to move from the electrolyte to the cavities, reversing on discharge. This charge-discharge cycle is known as a reduction/oxidation (i.e. redox) couple; for Ni(OH)2 one possible redox couple is cycling between the α/γ phases (see Figure 1).
A typical electro-active material is nickel hydroxide –Ni(OH)2 comprising a layered structure with intercalated water, forming gaps or cavities between the layers (see page image). When in contact with an electrolyte containing ions such as K+, Na+ or Li+ the application of a voltage charge causes the ions to move from the electrolyte to the cavities, reversing on discharge. This charge-discharge cycle is known as a reduction/oxidation (i.e. redox) couple; for Ni(OH)2 one possible redox couple is cycling between the α/γ phases (see Figure 1).
Fig. 1 Abstract graphic from 'Quantitative Resolution of Complex Stoichiometric Changes During Electrochemical Cycling by Density Functional Theory Assisted, Electrochemical Quartz Crystal Microbalance',
It is possible to gain some insight into degradation by using the experimental measurement technique Electrochemical Quartz Crystal Microbalance (EQCM). For Ni(OH)2, degradation leads to the formation of the new β-phase, (see Figure 1) and although EQCM measures changes of mass and charge, it provides no details of the redox reaction, and hence no information of the amount of intercalated ions, details that are important to a fundamental understanding of the mechanism behind degradation.
In a redox reaction where the final products are known, (e.g. different phases) and there are only two unknown stoichiometric coefficients involving the reactants, the two coefficients that balance the redox equation can be found from the change of mass and charge using linear algebra. In general, electro-active redox reactions contain more than two unknowns, which leads to an infinite number of stoichiometric solutions. By using experimental EQCM data it's possible to select a number of these solutions for the sets of coefficients, thereby defining the atomistic composition and stoichiometry of the unit cell of Ni(OH)2. This information is used to build atomistic models for computing with Density Functional Theory (DFT). The lowest calculated energy of formation identifies the structures for the redox reaction.
Novel findings
For the uncycled material in α-phase, DFT calculations predicted two possible structures with lowest formation energy, one where the cavities contained water (one water molecule per Ni(OH)2 unit) and one without (see Figure 2). At the time of the research, the literature described Ni(OH)2 as pristine material, free of intercalated species including water. The presence of intercalated water compatible with the DFT prediction was confirmed by the experimental team in Taiwan via thermogravimetric analysis (TGA), where the change in mass on heating demonstrated the material indeed contains water, and without it the material is prone to collapse.
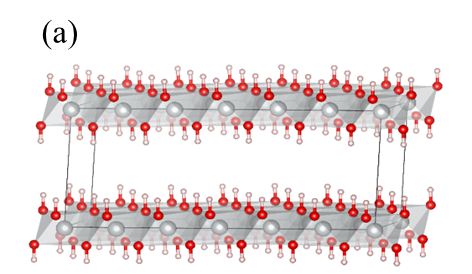
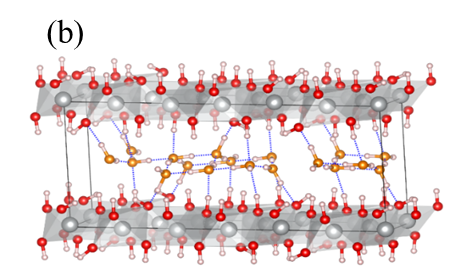
Fig. 2 Schematic representation of the computed optimized structures for (a) αNi(OH)2 free of water and (b) α-Ni(OH)2(H2O)δ=1. Ni: grey, O in NiO2: red, H: pink, O in H2O: orange. H-bonds are highlighted by means of dotted blue lines. The simulation cell (black continuous line) is also shown.
Experiments showed that when samples are cycled in electrolytes containing Li+ the electrode material degraded less than when cycled in solutions with Na+ and K+. This effect is demonstrated by
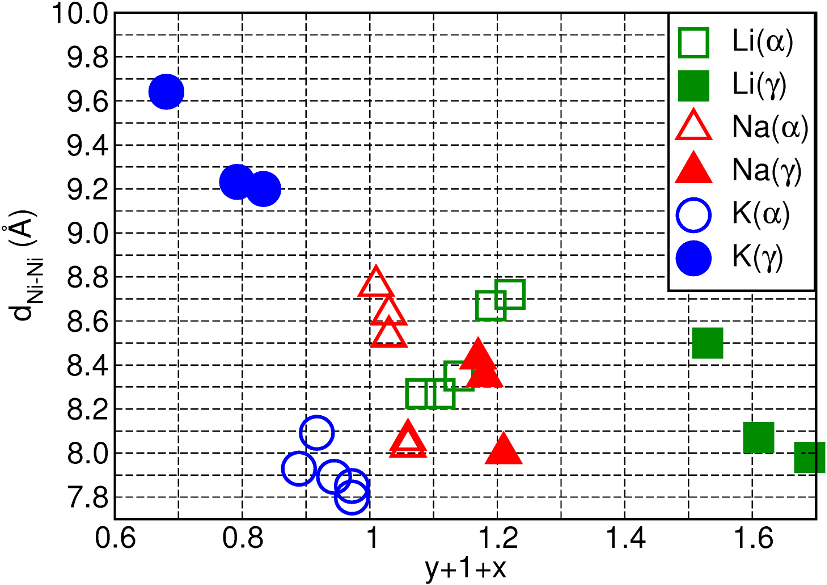
the computed average interlayer distances, where differences between phases α and γ are the largest for K+ (see Figure 3). Following six charge/discharge cycles a new EQCM signal appeared, indicating the formation of a new phase (β-phase) and accelerated degradation.
 The unique combination of EQCM and DFT simulations showed that when an ion enters the material cavity, the cavity-water is mostly removed in the presence of K+, to a lesser extent in the presence of Na+ and not at all in the presence of Li+. This suggests that during the removal of K+ (on discharge), if water molecules are not able to re-enter the host material fast enough, the structure will probably collapse, leading to the formation of a new phase and the electro-chemical degradation of the material. This phenomenon provides an explanation of the aforementioned new signal for K+ and Na+ after several charge/discharge cycles.
The unique combination of EQCM and DFT simulations showed that when an ion enters the material cavity, the cavity-water is mostly removed in the presence of K+, to a lesser extent in the presence of Na+ and not at all in the presence of Li+. This suggests that during the removal of K+ (on discharge), if water molecules are not able to re-enter the host material fast enough, the structure will probably collapse, leading to the formation of a new phase and the electro-chemical degradation of the material. This phenomenon provides an explanation of the aforementioned new signal for K+ and Na+ after several charge/discharge cycles.
Fig. 3 Cation-resolved analysis of the computed Ni-Ni interlayer distance (dNi-Ni, Å) as a function of the cumulative water (x) and cation (y) content (y+1+x) for the energy-favored optimized models of the charged (γ) and discharged (α) states, after electrochemical cycling.
Benefits
By combining disciplines from two different communities working in electronic structure theory and electrochemistry, this team of scientists developed a new DFT/experimental strategy - that adds to the tools of techniques available to explore the mechanism of ion-intercalation. This is a perfect example of a synergistic relationship between experiment and theory, and has laid the foundation for follow up work, supported by STFC's Ada Lovelace Centre. Begun in April 2020 in collaboration with experimental partners at Diamond Light Source (Dr Sofia Diaz-Moreno), Central Laser Facility (Dr Paul Donaldson), and STFC ISIS Neutron and Muon Source (Dr Daniel Bowron) the aim is to develop software to automate this novel approach, making it accessible to all interested users of STFC Facilities.
The paper 'Quantitative Resolution of Complex Stoichiometric Changes During Electrochemical Cycling by Density Functional Theory Assisted, Electrochemical Quartz Crystal Microbalance', is published in ACS Applied Energy Materials
Additional information about the DFT calculations
Approximately 100 models were built each comprising approximately 300 atoms; each formation energy calculation used on average 256 cores for about 24-36 hours. The calculations were performed on Supercomputing Wales, Archer, and Hartree Centre HPC. This research took 6 years to complete.